One way to analyse word relations is to examine their co-occurrence in the same context. This allows for the identification of potential semantic or lexical relationships between words. As previous studies showed word co-occurrences often reflect human stimuli-response pairs. In this paper significant sentence co-occurrences on word level were used to identify potential responses for word stimuli based on three automatically generated text corpora of the Leipzig Corpora Collection.
Epigenetics studies heritable phenotypic changes which are not due to changes in the DNA sequence. The molecular basis is chromatin forming a “beads on a string”-like structure of histones. Here we present a new tool called ChromatinVis to visualize ChIP-seq data. Before visualization, the histone modification data is segmented typically yielding several millions of data points. In our example, we process data from three cell types and three modifications resulting in eight combinations. The challenging problem is to study the global changes of histone modifications between different cell types. The data are clustered using the k means++ algorithm. For each cluster we allow the user to study the global and local distribution of histone marks using radial windmill charts. To analyze the configuration of the clusters in the data space we use scatterplots in combination with a Principle Component Analyses. A multitude of filtering options and several methods for outlier detection, like calculation of silhouette coefficients, allow the user to improve clustering. From a biological perspective, the tool gives a deeper insight into relationship between histone modifications.
Published as a poster at the Vizbi 2014 conference.

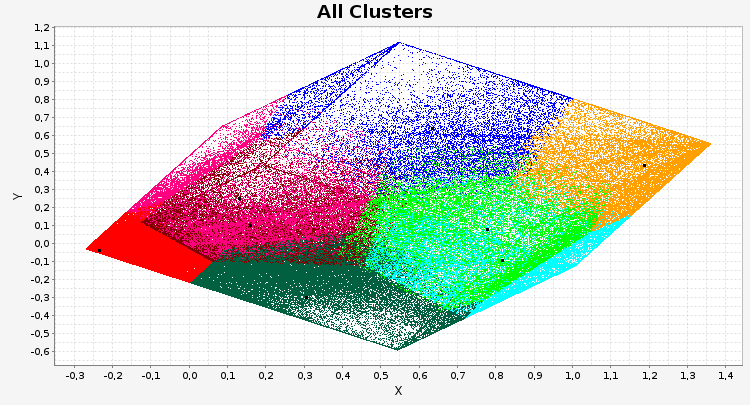
Over the last years, more and more biological data became available. Besides the pure amount of new data, also its dimensionality – the number of different attributes per data point – increased. Recently, especially the amount of data on chromatin and its modifications increased considerably. In the field of epigenetics, appropriate visualization tools designed for highlighting the different aspects of epigenetic data are currently not available. We present a tool called TiBi-Scatter enabling correlation analysis in 2D. This approach allows for analyzing multidimensional data while keeping the use of resources such as memory small. Thus, it is in particular applicable to large data sets.
TiBi-Scatter is a resource-friendly and easy to use tool that allows for the hypothesis-free analysis of large multidimensional biological data sets.
Published at the BioVis 2014