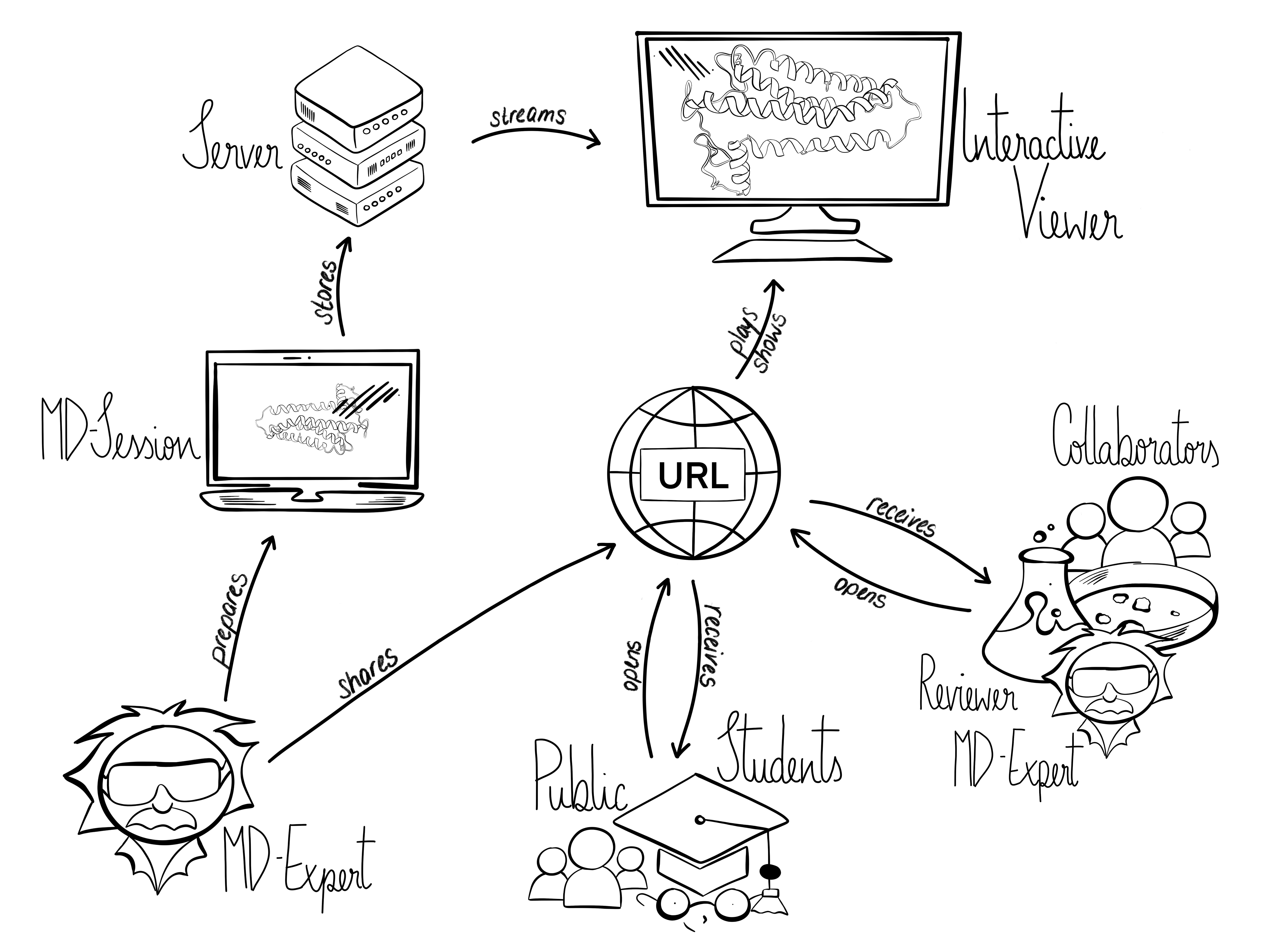
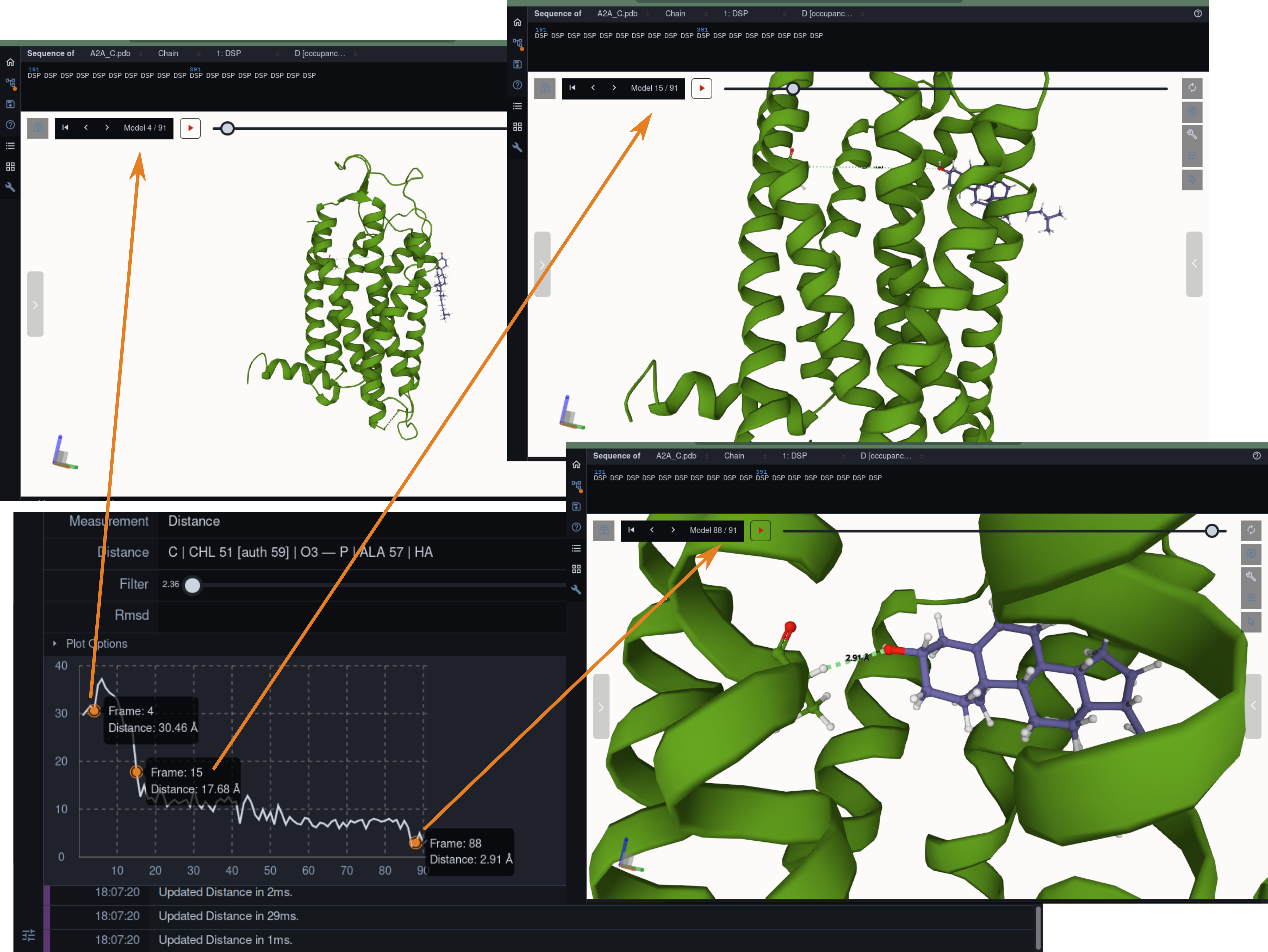
Molecular dynamics simulation is a proven technique for computing and visualizing the time-resolved motion of macromolecules at atomic resolution. The MDsrv is a tool that streams MD trajectories and displays them interactively in web browsers without requiring advanced skills, facilitating interactive exploration and collaborative visual analysis. We have now enhanced the MDsrv to further simplify the upload and sharing of MD trajectories and improve their online viewing and analysis. With the new instance, the MDsrv simplifies the creation of sessions, which allows the exchange of MD trajectories with preset representations and perspectives. An important innovation is that the MDsrv can now access and visualize trajectories from remote datasets, which greatly expands its applicability and use, as the data no longer needs to be accessible on a local server. In addition, initial analyses such as sequence or structure alignments, distance measurements, or RMSD calculations have been implemented, which optionally support visual analysis. Finally, based on Mol*, MDsrv now provides faster and more efficient visualization of even large trajectories compared to its predecessor tool NGL.

Background
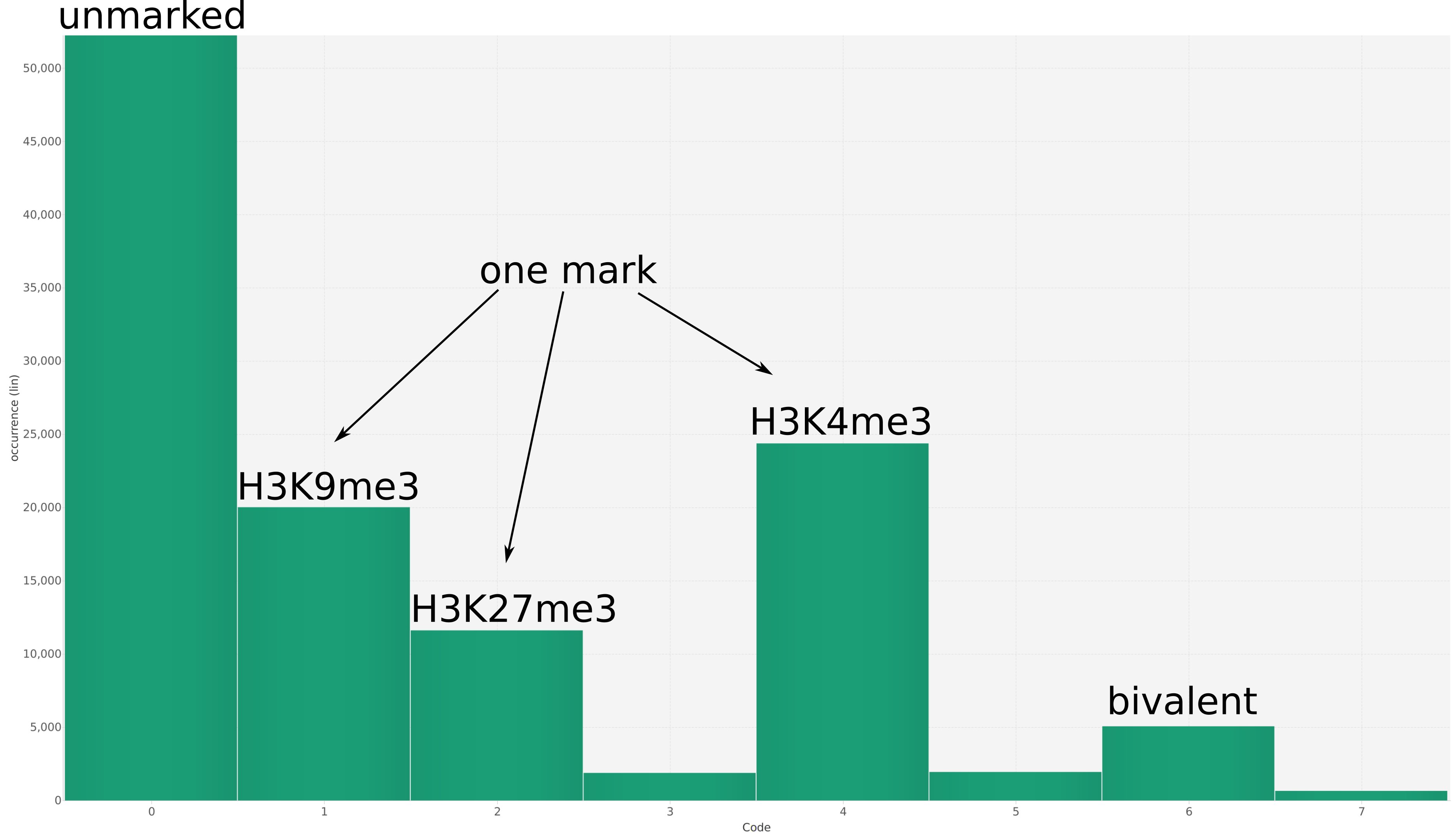
In epigenetics, the change of the combination of histone modifications at the same genomic location during cell differentiation is of great interest for understanding the function of these modifications and their combinations. Besides analyzing them locally for individual genomic locations or globally using correlations between different cells types, intermediate level analyses of these changes are of interest. More specifically, the different distributions of these combinations for different cell types, respectively, are compared to gain new insights.
Results and Discussion
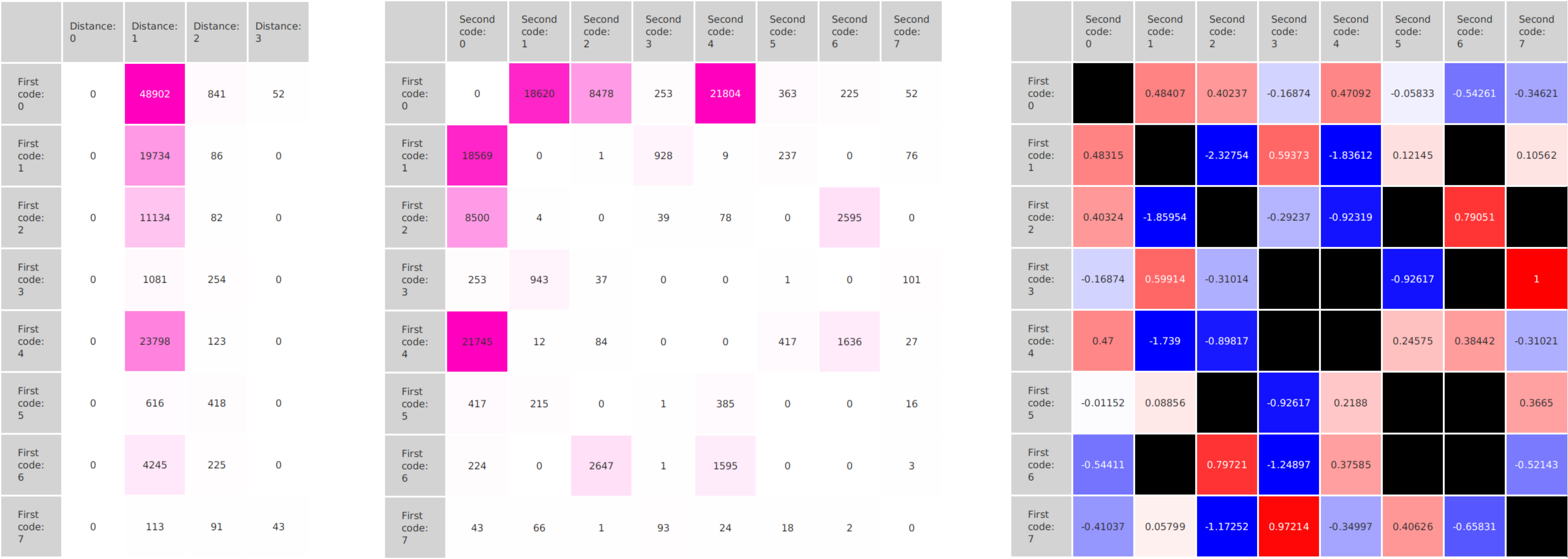
We propose a new tool called Masakari that allows segmenting genomes based on lists of ranges having a certain property, e.g., peaks describing histone modifications. It provides a graphical user interface allowing to select all data sets and setting all parameters needed for the segmentation process. Moreover, the graphical user interface provides statistical graphics allowing to assess the quality and suitability of the segmentation and the selected data.
Conclusion
Masakari provides statistics based visualizations and thus fosters insights into the combination of histone modification marks on genome ranges, and the differences of the distribution of these combinations between different cell types.

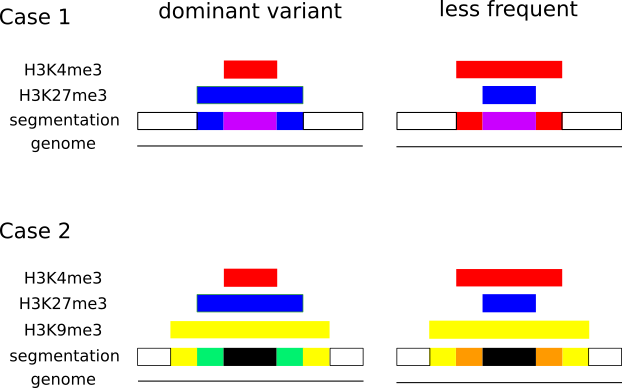

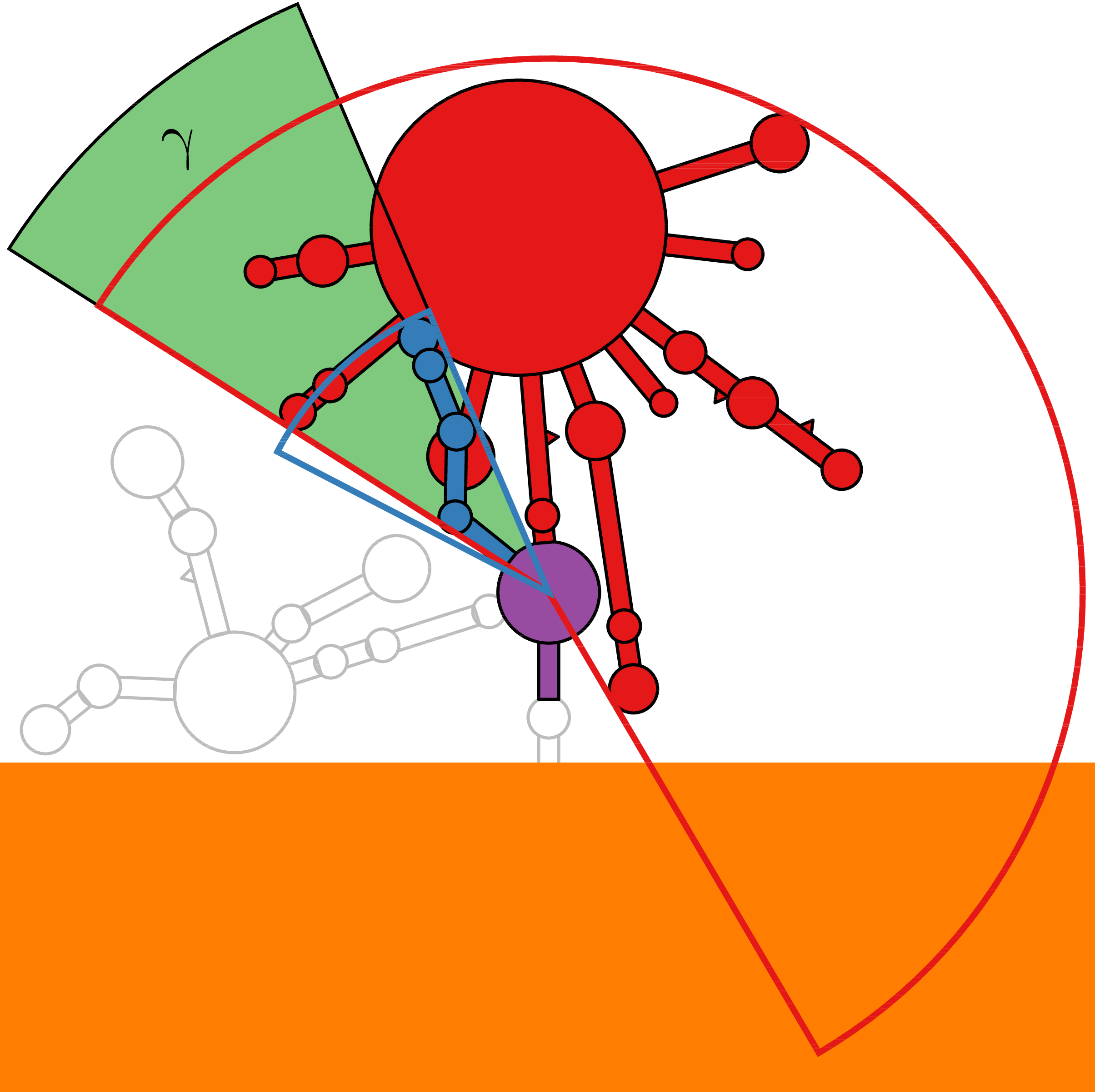
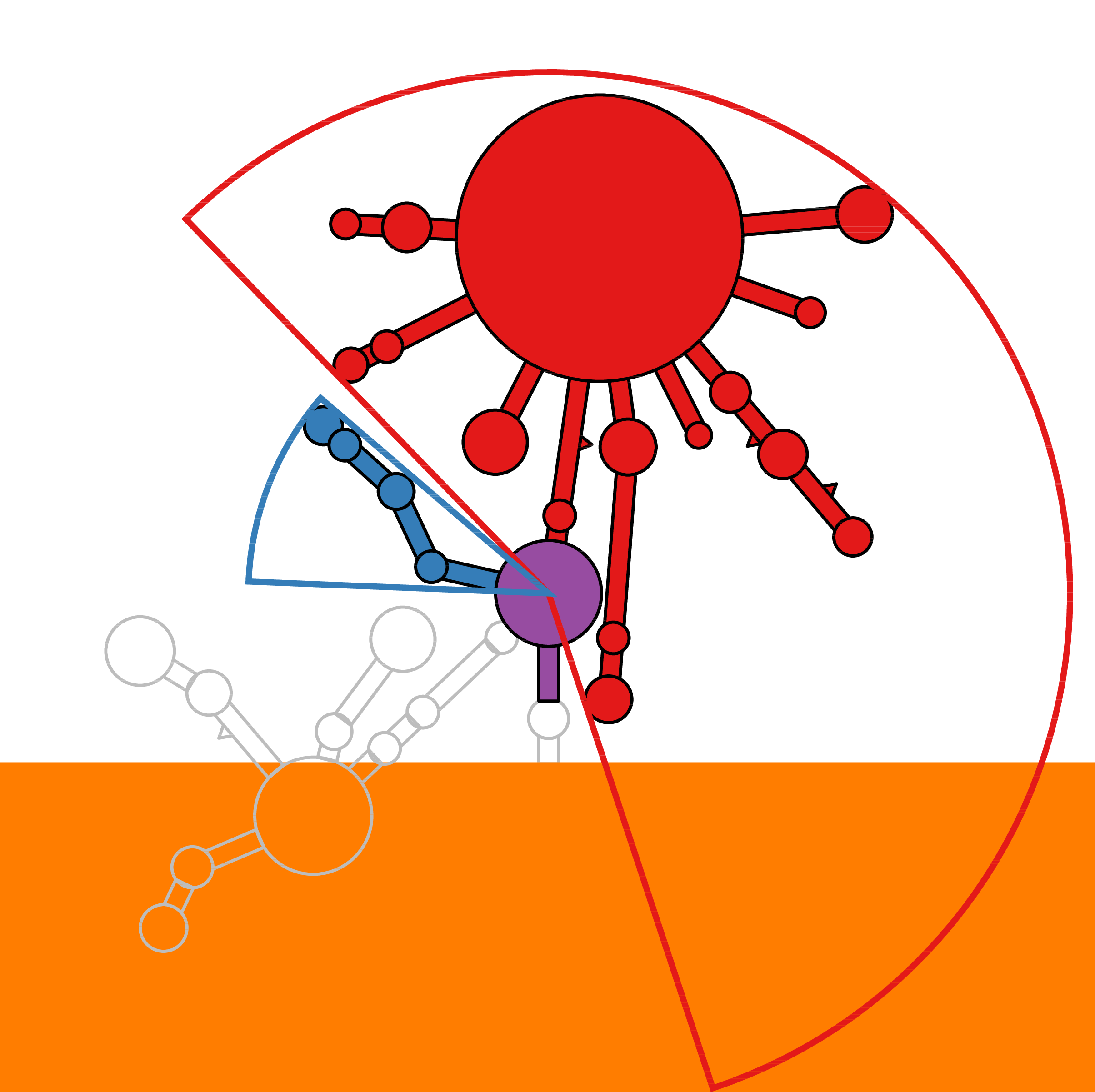
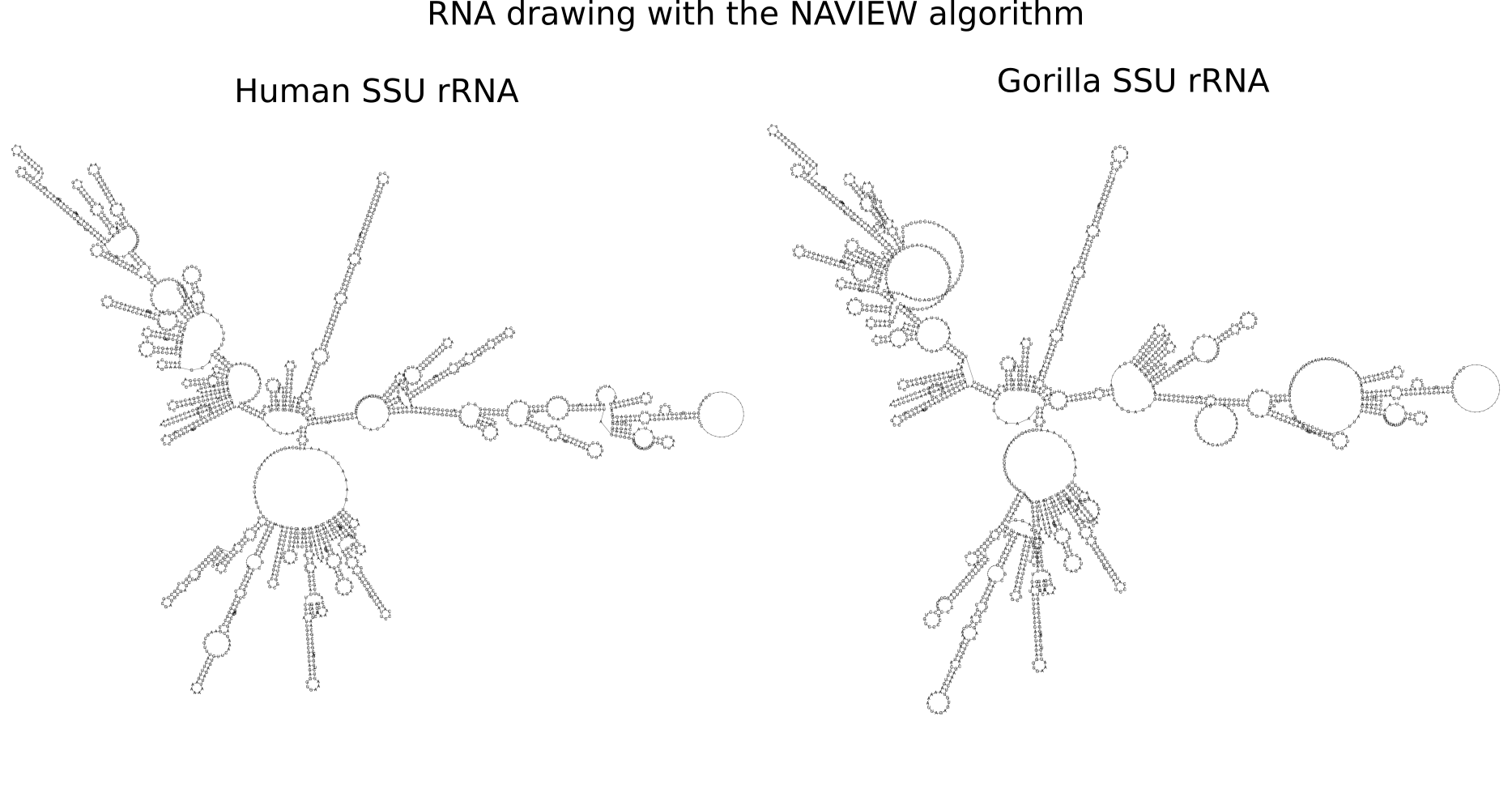
RNA secondary structure is a useful representation for studying the function of RNA, which captures most of the free energy of RNA folding. Using empirically determined energy parameters, secondary structures of nucleic acids can be efficiently computed by recursive algorithms. Several software packages supporting this task are readily available. As RNA secondary structures are outerplanar graphs, they can be drawn without intersection in the plane. Interpretation by the practitioner is eased when these drawings conform to a series of additional constraints beyond outerplanarity. These constraints are the reason why RNA drawing is difficult. Many RNA drawing algorithms therefore do not always produce intersection-free (outerplanar) drawings.
To remedy this shortcoming, we propose here the RNApuzzler algorithm, which is guaranteed to produce intersection-free drawings. It is based on a drawing algorithm respecting constraints based on nucleotide distances (RNAturtle). We investigate relaxations of these constraints allowing for intersection- free drawings. Based on these relaxations, we implemented a fully automated, simple, and robust algorithm that produces aesthetic drawings adhering to previously established guidelines. We tested our algorithm using the RFAM database and found that we can compute intersection-free drawings of all RNAs therein efficiently.
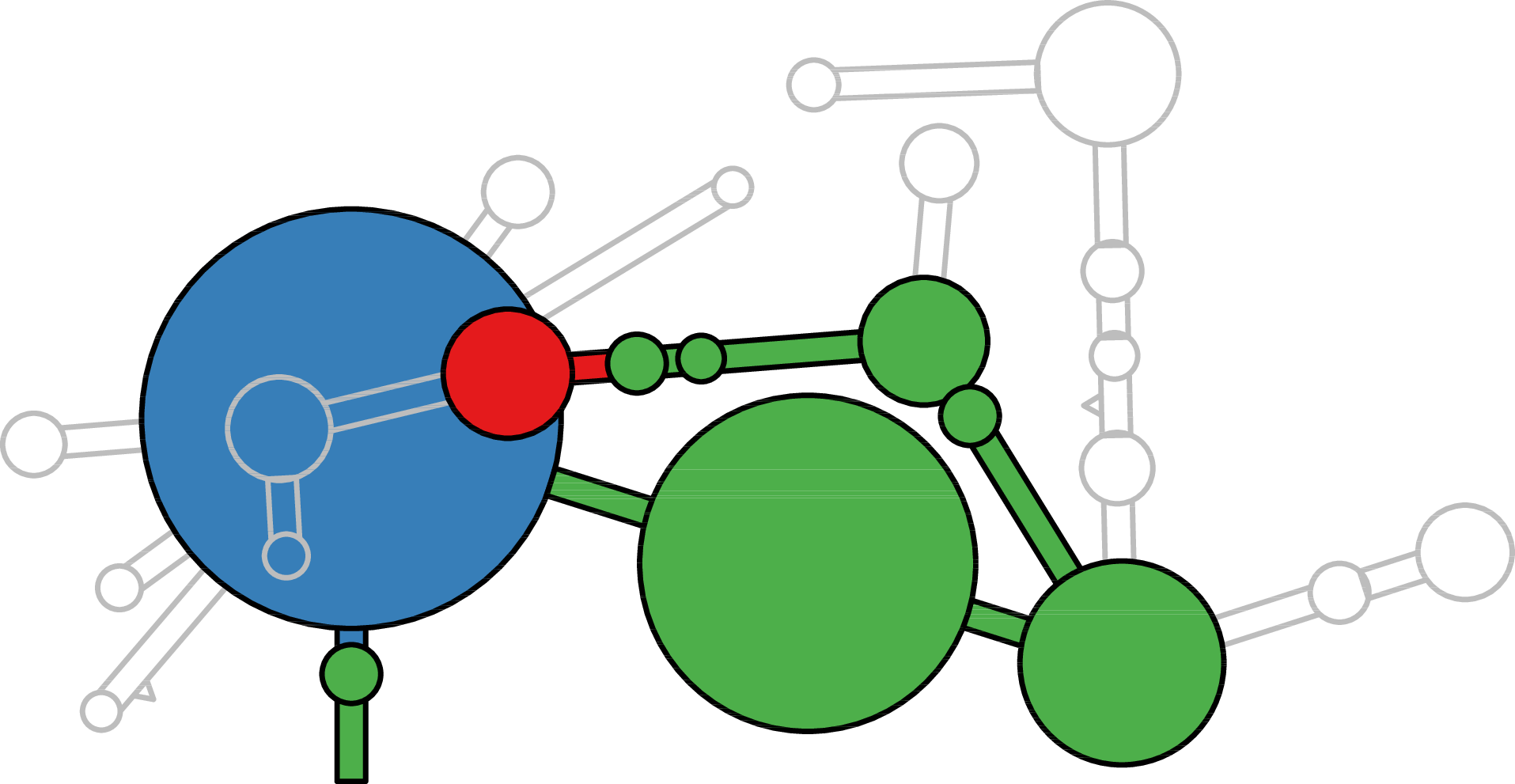
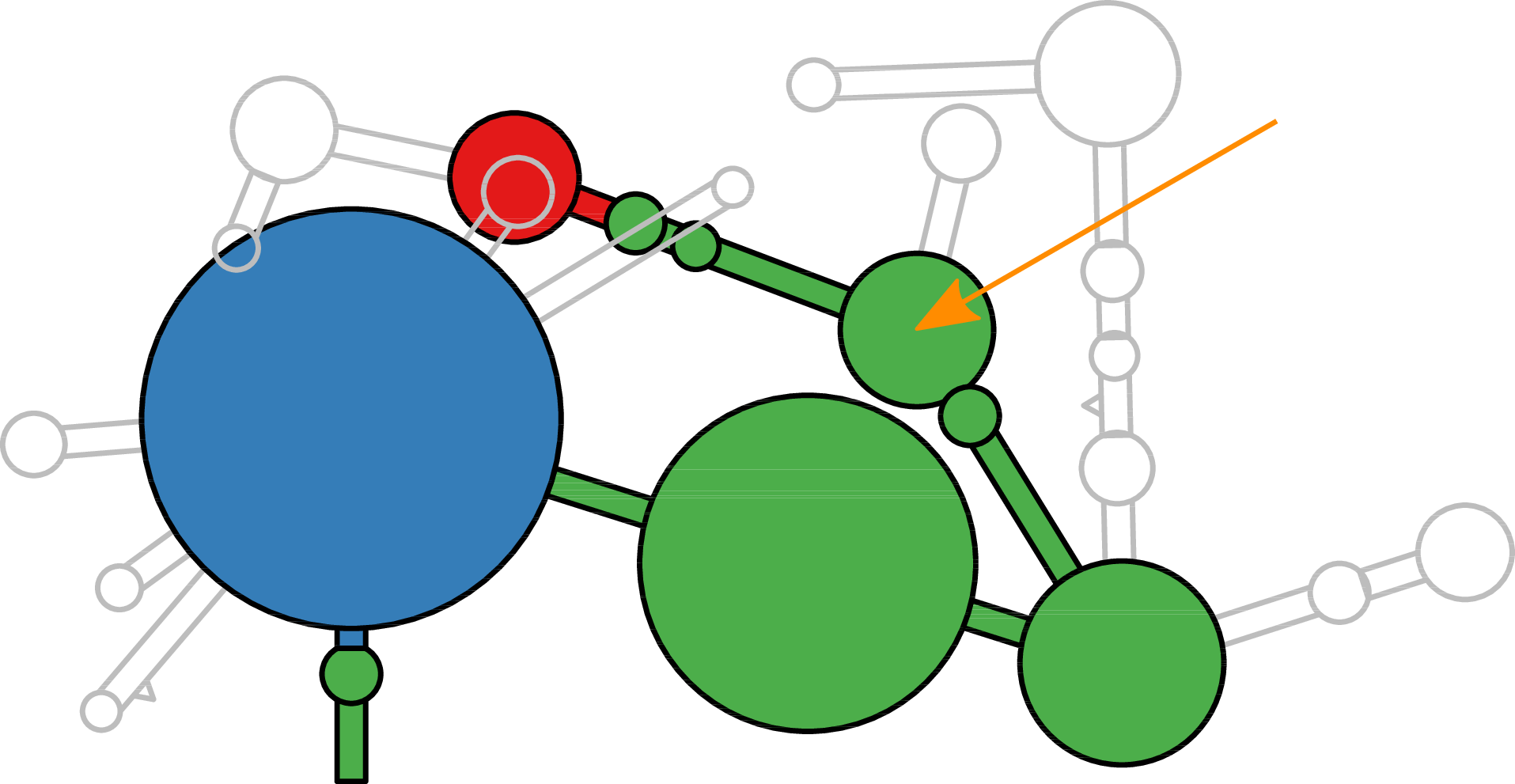

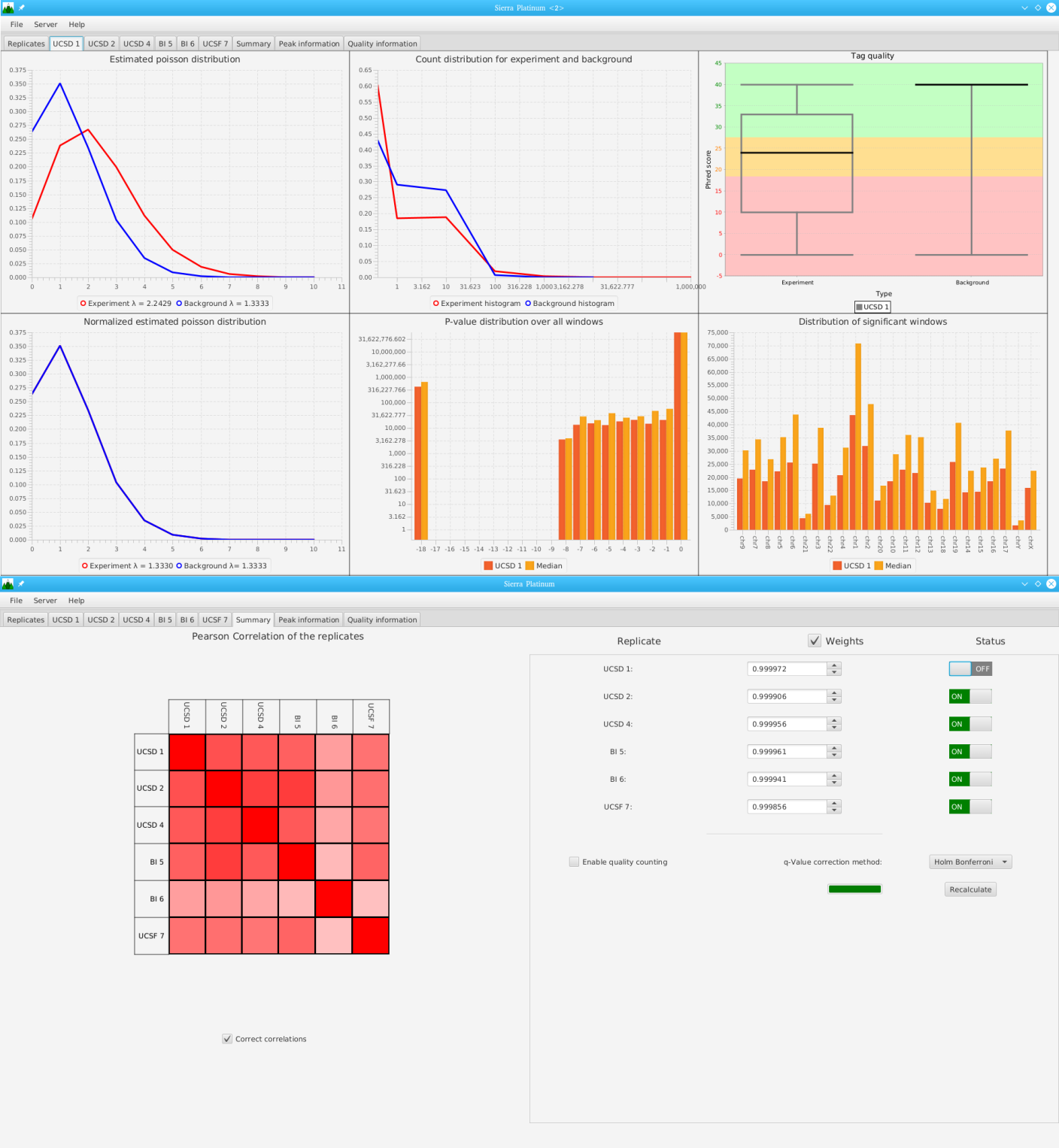
Sierra Platinum is a fast and robust peak-caller for replicated ChIP-seq experiments with visual quality-control and -steering. The required computing resources are optimized but still may exceed the resources available to researchers at biological research institutes. Sierra Platinum Service provides the full functionality of Sierra Platinum: using a web interface, a new instance of the service can be generated. Then experimental data is uploaded and the computation of the peaks is started. Upon completion, the results can be inspected interactively and then downloaded for further analysis, at which point the service terminates.

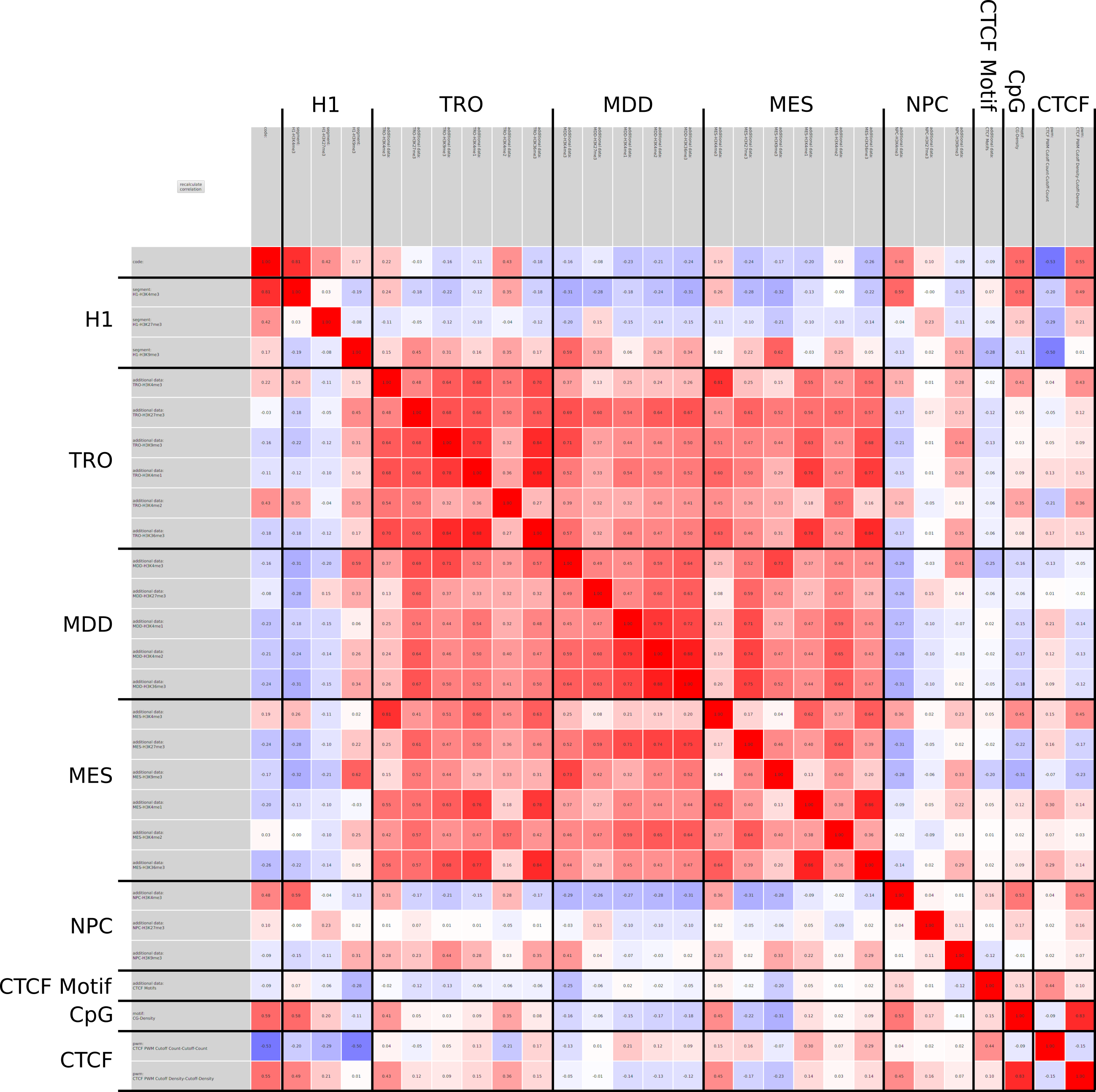
A major goal in epigenetics is understanding how cells differentiate into different cell types. Besides the increase of individual data sets, the amount of replicated experiments generating a tremendous amount of data is ever increasing. While biologists primarily analyze their data on the highest level using statistical correlations or on the lowest level analyzing nucleotide sequences, determining the fate of histone modifications during cell specification necessitates improved analysis capabilities on one or more intermediate levels. For this type of analysis, it proved to be very useful to use tiled binned scatter plot matrices showing binary relationships or to use tiled binned 3D scatter plots showing ternary relationships. Quarternary or general n-ary relationships are not easily analyzable using visualization techniques like scatter plots, only. Therefore, we augmented existing clustering methods with the tiling and binning idea enabling the analysis of n-ary relationships. Analyzing the changes of histone modifications comparing two cell lines using tiled binned clustering, we found new, unknown relations in the data.

