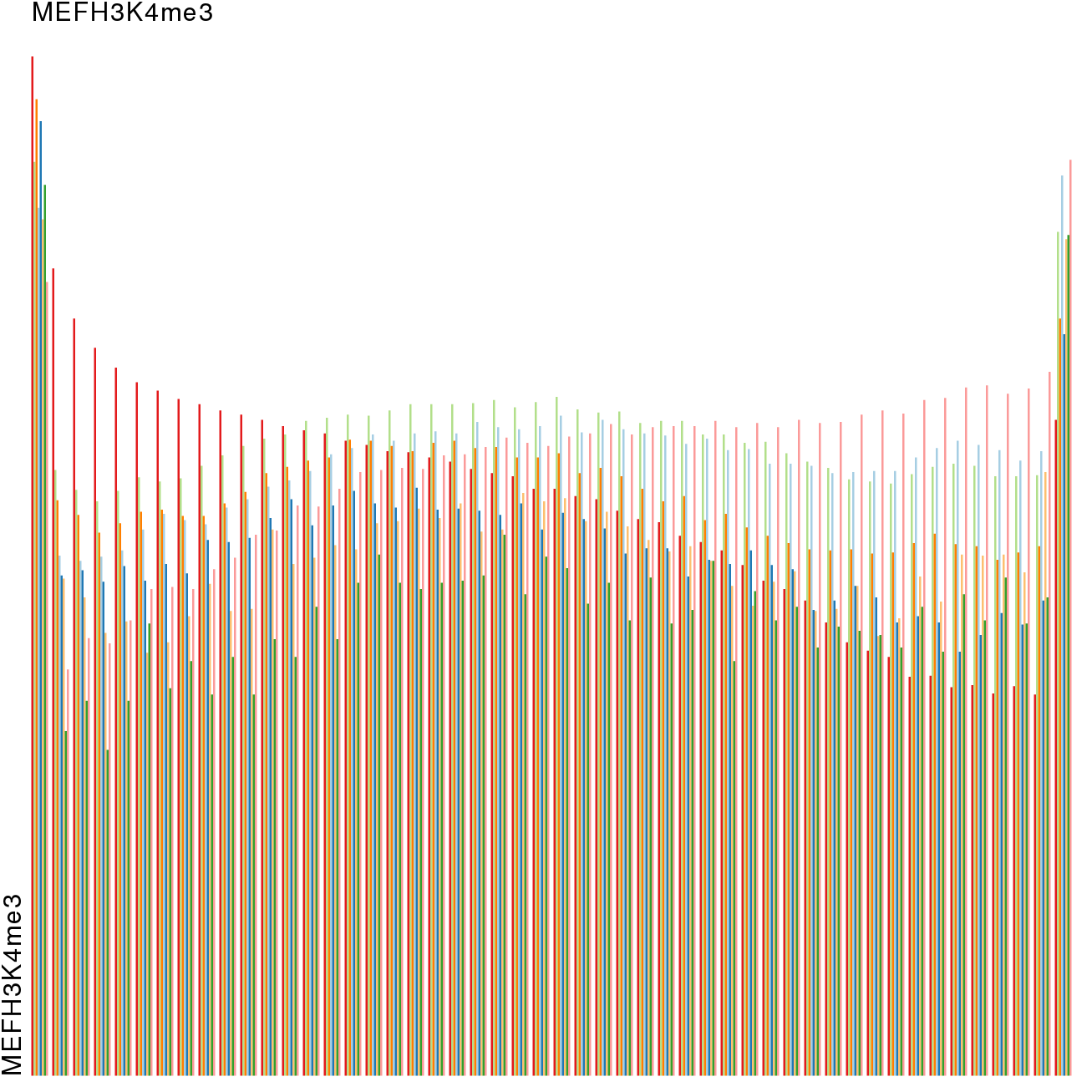
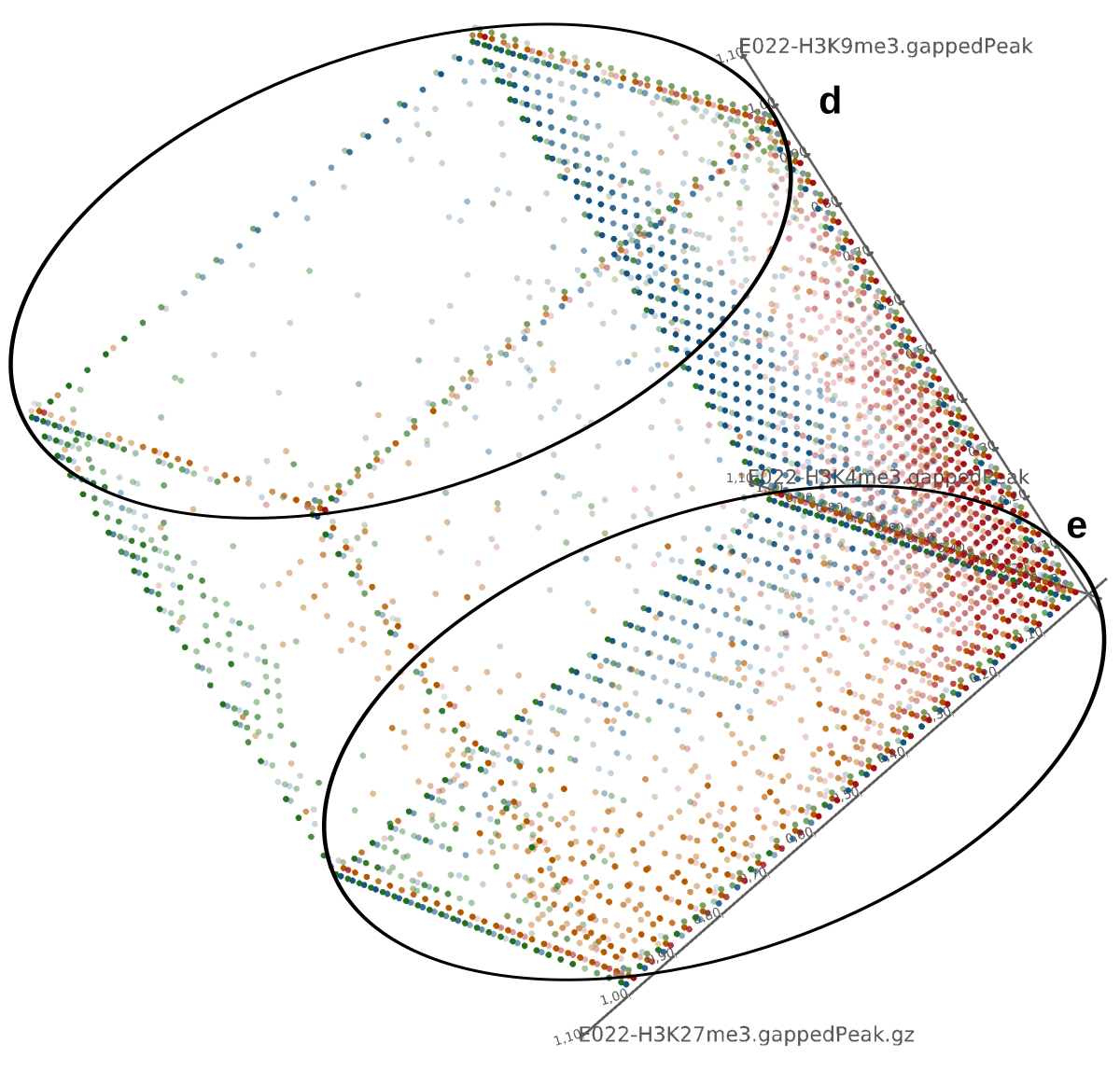
Epigenetics data is very important for understand- ing the differentiation of cells into different cell types. More- over, the amount of epigenetic data available was and still is considerably increasing. To cope with this big amount of data, statistical or visual analysis is used. Usually, biologists analyze epigenetic data using statistical methods like correlations on a high level. However, this does not allow to analyze the fate of histone modifications in detail during cell specification or to compare histone modifications in different cell lines. Tiled binned scatter plot matrices proved to be very useful for this type of analysis showing binary relationships. We adapted the idea of tiling and binning scatter plots from 2D to 3D, such that ternary relationships can be depicted. Comparing tiled binned 3D scatter plots—the new method—to tiled binned 2D scatter plot matrices showed, that many relations that are difficult or impossible to find using tiled binned 2D scatter plot matrices can easily be observed using the new approach. We found that using our approach, changes in the distribution of the marks over time (different cell types) or differences between different replicates of the same cell sample are easy to detect. Tiled binned 3D scatter plots proved superior compared to the previously used method due to the reduced amount of overplotting leading to less interaction necessary for gaining similar insights.

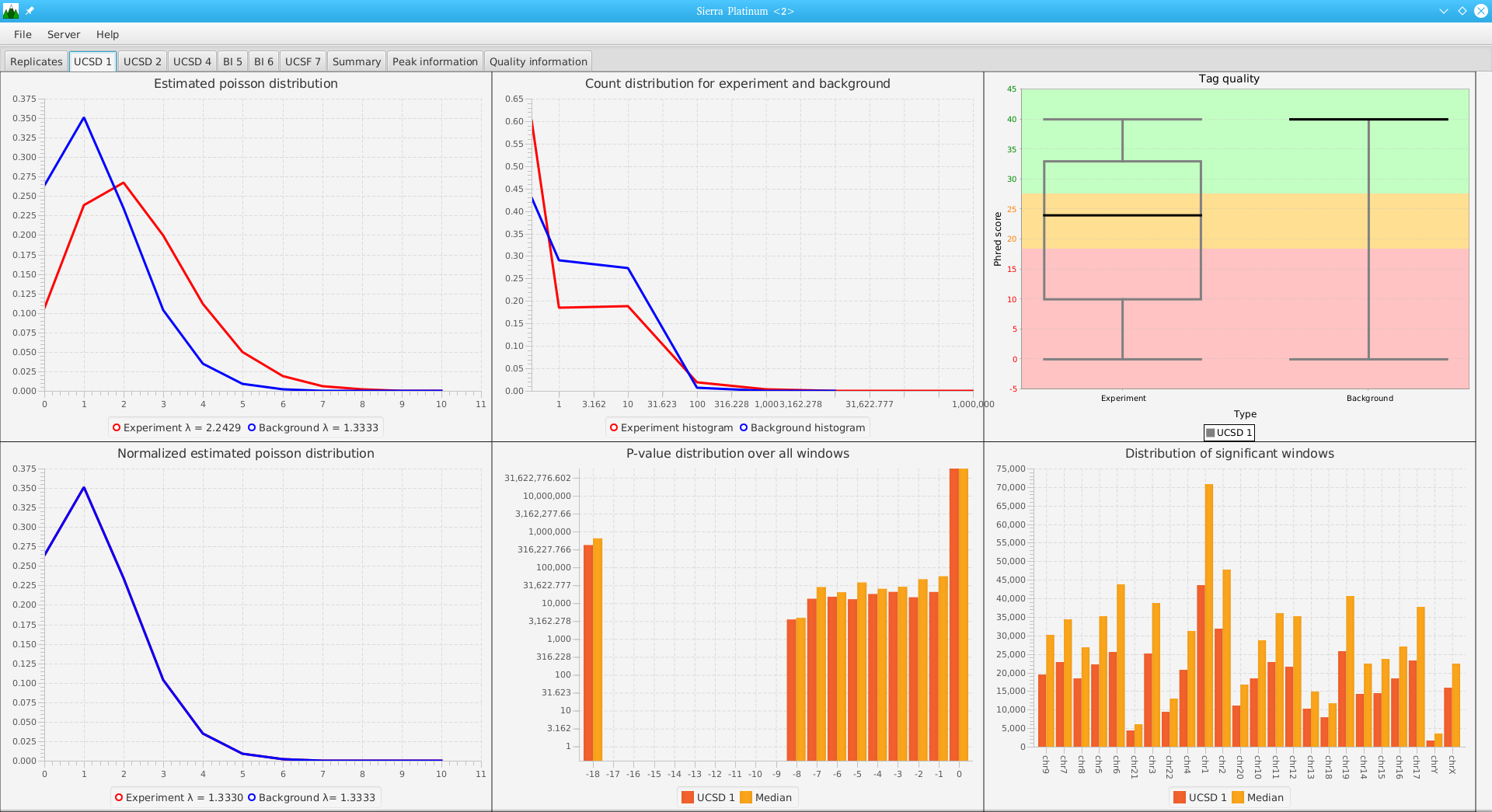
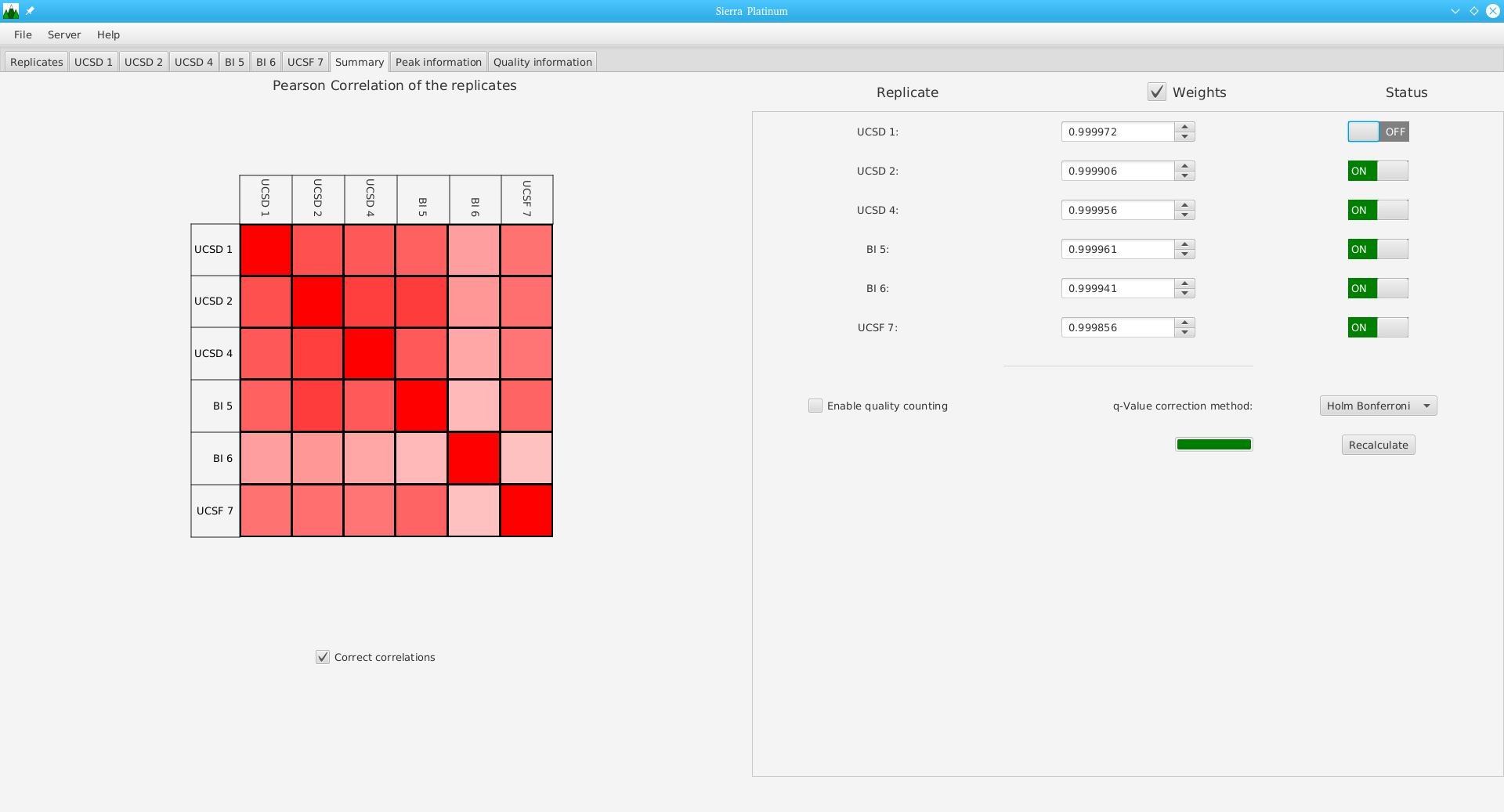
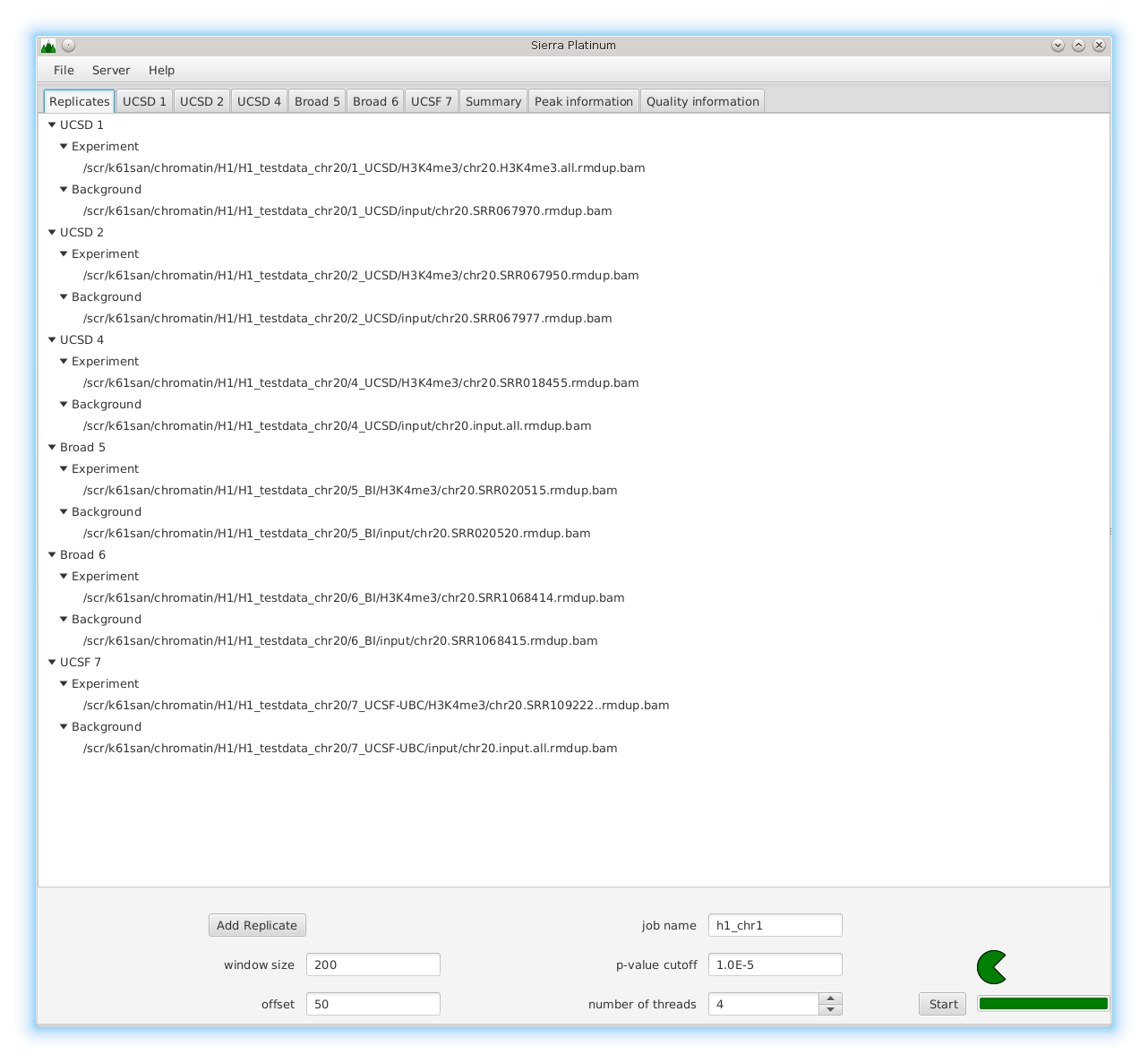
DNA bound proteins such as transcription factors and modified histone proteins play an important role in gene regulation. Therefore, their genomic locations are of great interest. Usually, the location is measured using ChIP-seq and analyzed using a peak-caller. While replicated ChIP-seq experiments become more and more available, they are still mostly analyzed using methods based on peak-callers for single replicates. The only exception is PePr, which allows peak calling of several replicates. However, PePr does not provide quality measures to assess the result of the peak-calling process. Moreover, its underlying model might not be suitable for the conditions under which the experiments are performed. We propose a new peak-caller called `Sierra Platinum' that not only allows to call peaks for several replicates but also provides a variety of quality measures. Together with integrated visualizations, the quality measures support the assessment of the replicates and the resulting peaks. We show that Sierra Platinum outperforms methods based on single-replicate peak-callers as well as PePr using a newly generated benchmark data set and using real data from the NIH Roadmap Epigenomics Project.
Cognitive abilities, such as memory, learning, language, problem solving, and planning, involve the frontal lobe and other brain areas. Not much is known yet about the molecular basis of cognitive abilities, but it seems clear that cognitive abilities are determined by the interplay of many genes. One approach for analyzing the genetic networks involved in cognitive functions is to study the coexpression networks of genes with known importance for proper cognitive functions, such as genes that have been associated with cognitive disorders like intellectual disability (ID) or autism spectrum disorders (ASD). Because many of these genes are gene regulatory factors (GRFs) we aimed to provide insights into the gene regulatory networks active in the human frontal lobe. Using genome wide human frontal lobe expression data from 10 independent data sets, we first derived 10 individual coexpression networks for all GRFs including their potential target genes. We observed a high level of variability among these 10 independently derived networks, pointing out that relying on results from a single study can only provide limited biological insights. To instead focus on the most confident information from these 10 networks we developed a method for integrating such independently derived networks into a consensus network. This consensus network revealed robust GRF interactions that are conserved across the frontal lobes of different healthy human individuals. Within this network, we detected a strong central module that is enriched for 166 GRFs known to be involved in brain development and/or cognitive disorders. Interestingly, several hubs of the consensus network encode for GRFs that have not yet been associated with brain functions. Their central role in the network suggests them as excellent new candidates for playing an essential role in the regulatory network of the human frontal lobe, which should be investigated in future studies.
One way to analyse word relations is to examine their co-occurrence in the same context. This allows for the identification of potential semantic or lexical relationships between words. As previous studies showed word co-occurrences often reflect human stimuli-response pairs. In this paper significant sentence co-occurrences on word level were used to identify potential responses for word stimuli based on three automatically generated text corpora of the Leipzig Corpora Collection.
Over the last years, more and more biological data became available. Besides the pure amount of new data, also its dimensionality – the number of different attributes per data point – increased. Recently, especially the amount of data on chromatin and its modifications increased considerably. In the field of epigenetics, appropriate visualization tools designed for highlighting the different aspects of epigenetic data are currently not available. We present a tool called TiBi-Scatter enabling correlation analysis in 2D. This approach allows for analyzing multidimensional data while keeping the use of resources such as memory small. Thus, it is in particular applicable to large data sets.
TiBi-Scatter is a resource-friendly and easy to use tool that allows for the hypothesis-free analysis of large multidimensional biological data sets.
Published at the BioVis 2014